 MOLDEN
MOLDEN
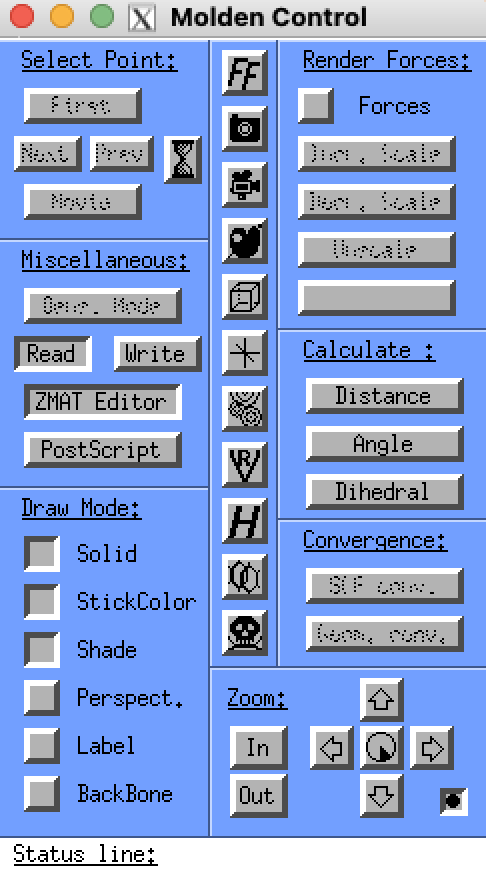
MOLDEN is a software which can create, edit, and optimize structures of molecules for their implementation into PACKMOL. To use MOLDEN, the following steps must be completed:
Download XQuartz from their website.
Login to the cluster with your KUID (see Cluster Commands).
module load molden/7.3Enter the directory where you wish to run MOLDEN. It is recommended that for reading accepted files for Z-Matrix editing to be within directories that contain the file(s) of interest.
Type
moldento start your MOLDEN session.
Once MOLDEN is opened, the Control Panel screen will appear alongside the visualization screen (see right). The MOLDEN Control panel will allow you to read files, edit your Z-matrix, calculate molecular parameters, and optimize the structure of a molecule. These commonly used features will be explained below; for more details on how to use MOLDEN to create molecules, see the Developing Molecules for LAMMPS section. To quit MOLDEN, press the skull image in the MOLDEN Control Panel. This will close all tabs in your MOLDEN session and return you to the terminal.
Reading Files and Editing the Z-Matrix
MOLDEN allows the ability for files to be read into the Z-matrix editor for quick creation of molecules. This can be done via selecting the “Read” button within the “Miscellaneous” header on the control panel. The file can then be selected from the window. Once the file is read, the Z-matrix can be opened within the “ZMAT Editor” under the “Miscellaneous” header. The Z-matrix provides key details for the bonds, angles, and dihedrals of the system. This can be viewed for editing the parameters as well as editing the molecule itself.
Note
When atoms are opened, it is helpful to select the “Solids; Sticks” buttons under the “Draw Mode” header to see the molecule clearly.

Z-Matrix Basics
If you are unfamiliar with Z-matrices and how to read them within MOLDEN, fear not! Z-matrices provide all the necessary details to form the molecule and make sure its atoms are oriented properly.
Following the Z-matrix image, the first column displays the specific atom within the molecule. The first row shows the first atom in the molecule (which may not necessarily be the central atom!). This row is known as Row 1 and will be important when identifying bonds, angles, and dihedrals. As you progress down to the next row, a new column is added. This describes a bond between the atom in Row 1 and the atom in Row 2. The numbed columns between each parameter denote which atom is bonded to which; the third atom which composes the angle; and the final fourth atom that composes the dihedral. These values cannot be repeated within the specific row and are chosen carefully to account for as much of the molecule as possible. From the Z-matrix image, the visualization screen can correlate which atoms are affiliated with which parameter.
Note
It is highly recommended to find molecule files online which can be imported to MOLDEN to auto-create Z-matrices by reading their files. For example, a .xyz or .mol2 file can be read into MOLDEN if it is uploaded onto the cluster. As shown with the example, the first atom is not necessarily the central atom and must be chosen carefully to ensure the molecule is created correctly.
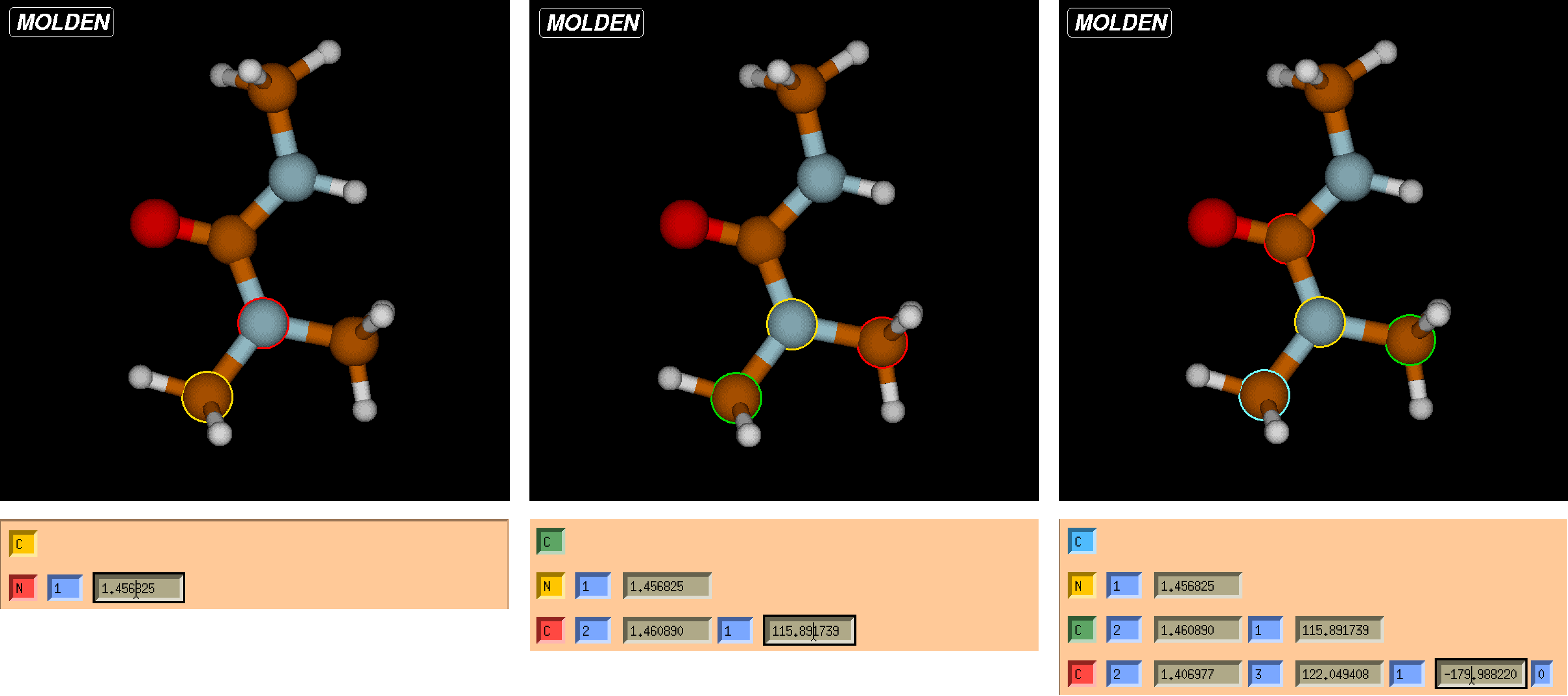
From the Z-matrix, it can be shown that the selected atom is colored red, and its secondary bonded atom is in yellow. You can also see this as both of these atoms are colored in the visualization screen. As the next row is added, a new column for angle is added. This colors the atoms of the molecule similar to that for bonds, but with a third atom colored green. The central atom will always be yellow when viewing angles. Lastly, the final column added is for that of dihedrals. These can be seen with the central atom still in yellow, but with a final fourth atom outlined in blue.
Creating, Editing, and Optimizing Structures
Creating molecules is possible within MOLDEN by a similar process to viewing the Z-matrix itself. You can select the “ZMAT Editor” from the MOLDEN Control panel to open an empty Z-Matrix. Then, you can press the “Add Line” button to add atoms to the system. As more lines are added, you must select other atoms to show bonds, angles, and dihedrals for the system. These can be edited later but must be initially stated in order for the atom to be added. Selecting bonded atoms can be done either by clicking lines in the Z-matrix or clicking atoms directly in the visualization screen.
Editing molecules can be done simply by clicking the data values for bond/angle/dihedral and typing in new values to edit. Pressing enter will update the structure, or you can also press “Apply Changes to Current Z-Mat” to update the structure. There are times where adjusting a value may cause the structure to become unstable, and MOLDEN will let you know within the terminal and also within the Status Line on the MOLDEN Control Panel. If you are attempting to update the matrix and nothing changes, look at these places for potential warnings that prevent an unstable structure from forming.
Optimizing a structure can be done by simply pressing the “FF” button on the MOLDEN Control Panel. You can use “Tinker CHARMM” to optimize the structure. This will automatically find the most stable configuration for the molecule and will update its parameters accordingly. From this, you can enter a file name and export the coordinates (select the “Cartesian” button and “xyz” from the bottom of the Z-matrix editor panel) or export the Z-matrix itself (select the “Gaussian” button and enter a file name) by hitting the “Write Z-Matrix” button. See below for details.
Note
There may be issues optimizing the structure for new users. You can instead follow the instructions in the Developing Molecules for LAMMPS to optimize the geometry in another way (via LAMMPS) if MOLDEN tells you that you have “untyped atoms” in the system. You can also use Avogadro to create and optimize Z-matrices if you are familiar.
Saving Z-Matrices and Coordinates
Editing a structure in MOLDEN does not automatically update the read files, changes must be saved and exported for permanent changes. This can be done simply by pressing the Gaussian or Cartesian buttons at the bottom of the Z-matrix editor to export Z-matrices or coordinates, respectively. For either case, a file name must be specified within the text box above the selection buttons. For example, an xyz file that is exported will be shown in the directory as the following:
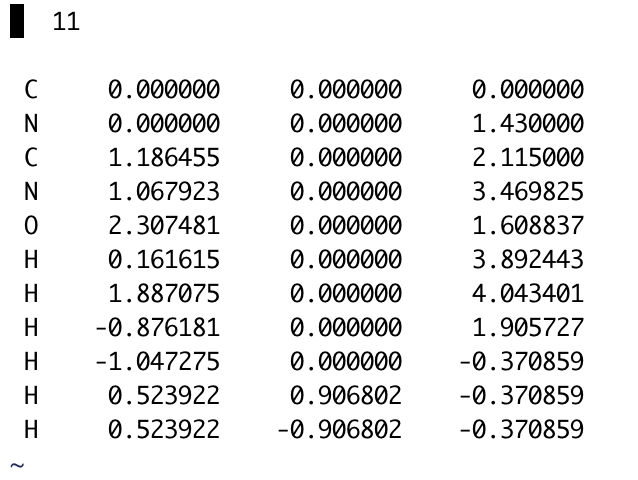
When creating a molecule for LAMMPS ( Developing Molecules for LAMMPS), the Cartesian coordinates must be used for PACKMOL. In this case, .xyz files are the easiest way to quickly add these to the necessary files. This can be done by selecting the “Cartesian” button and then pressing the “xyz” file from the selection menu. Make sure to press “Write Z-Matrix” to export the files accordingly!
Gaussian exports will save the Z-Matrix data for the desired file name. This can be done if you have plans to edit the Z-Matrix later, or plans to view the structure at some other time. It is recommended that in the case of developing molecules for LAMMPS that both the Z-matrix and the Cartesian coordinates are saved to ensure the molecule is still available should any issues occur during the PACKMOL process.