 PACKMOL
PACKMOL
PACKMOL creates the necessary pieces for a LAMMPS simulation data file. Users input the relevant force field data and PACKMOL creates the Cartesian system of atom coordinates and formats their relevant force field parameters in a LAMMPS-friendly format.
Note
This page is adapted from the My Code Collection PACKMOL documents, written by Dr. Zeke A. Piskulich.
Note
Details on how to create molecules for PACKMOL can be found within the Developing Molecules for LAMMPS and MOLDEN docs.
Installing PACKMOL
To install and successfully run PACKMOL, it is necessary to download the relevant materials to your cluster profile.
Download the codes from Zeke’s GitHub:
Choose somewhere safe to keep the codes, such as your
home/orwork/directory.Type the following command when you are within the desired location:
git clone https://github.com/piskuliche/My-Code-Collection.git
Preparing PACKMOL
Change the path in the
make.shfile to the location of theMy-Code-Collection/directory.Place the files from
module/to some private directory. This can be done by simply making another directory, known asprivatemodules/within the samehome/orwork/directory as where you placed theMy-Code-Collection/codes.Within the
privatemodules/directory, edit each.luafile and make the following edits:Change the path to the following:
/pwd/My-Code-Collection/bin/file/wherepwdis the path to theMy-Code-Collection/contents.Remove each line that loads python from all
.luafiles.
Return to your default cluster page (i.e. type
cd) and edit your bash profile:vi .bash_profile. Within it, type the following:module use /owd/private_mod_dir_namewherepwdis the path to theprivatemodulesdirectory.Source the profile
source .bash_profile(and logout/in).Edit the
build.pycode by completing the following steps:cd My-Code-Collection/bin/build/vi build.pyChange line 606 to:
subprocess.call(["/kuhpc/work/thompson/e497b540/packmol/packmol < system.pmol > packmol.output"], shell=True).
When you are ready to use
build.py, you must typemodule load build. It is recommended that you do this within your bash profile if you are routinely building PACKMOL contents. This can be done similarly to the edits for the bash profile in Step 1.
Preparing the PACKMOL Contents
PACKMOL input files are incredibly detailed and specific in order to yield a desired molecule. Unlike LAMMPS, PACKMOL insists on every discrete parameter as well as every type of parameter to be listed from a given set of force field contents. Details on the format and structure of the relevant PACKMOL files are shown blow.
Create a connectivity file (similar in appearance to a LAMMPS data file) with the name
molecule.connectwheremoleculeis the name of the species you are creating. Edit the file as follows:# HEADER (optional) # atoms # bonds # angles # dihedrals # impropers Coords # ALL coordinates must be shown, for all three dimensions, for ALL atoms in the system. 1 x y z 2 x y z N x y z # where N is the number of atoms in the molecule Types # ALL types must be shown, for ALL atoms in the system. 1 atom_number 2 atom_number N atom_number # show ALL atoms and their associated types. EVERY atom must be accounted for, where N is the number of atoms in the molecule. Masses # ALL masses must be shown, for ALL atoms in the system. 1 atom_mass 2 atom_mass N atom_mass # show ALL atoms and their associated masses, for 1-N atoms. Bonds # ALL bonds must be shown, for ALL atoms in the system. 1 bond_type_number atom1 atom2 2 bond_type_number atom1 atom2 NB bond_type_number atom1 atom2 # show ALL bonds with their associated bond type number and associated atoms. Angles # ALL angles must be shown, for ALL atoms in the system. 1 angle_type_number atom1 atom2 atom3 2 angle_type_number atom1 atom2 atom3 # atom2 is the central atom for the angle NA angle_type_number atom1 atom2 atom3 # show ALL angles with their associated angle type number and atoms. Dihedrals # ALL dihedrals must be shown, for ALL atoms in the system 1 dihedral_type_num atom1 atom2 atom3 atom4 2 dihedral_type_num atom1 atom2 atom3 atom4 # where atom2 and atom3 are the central atoms ND dihedral_type_num atom1 atom2 atom3 atom4 # show ALL dihedrals with their associated type number and atoms. Impropers 1 improper_type_num atom1 atom2 atom3 atom4 2 improper_type_num atom1 atom2 atom3 atom4 # where atom1 is the central atom NI improper_type_num atom1 atom2 atom3 atom4 # show all relevant impropers (by this point all other contributions should relieve you from finding all impropers) with their associated type number and atoms.
Note
All headers for
Coords,Types,Bonds, etc. require a blank line prior and following for correct formatting. For molecules that do not contain the aforementioned qualities can have their associated sections blank. Molecules without any aforementioned qualities do not need associated files (see below). The sections which do not apply to the molecule being built can be ignored. However, it is important to read the Examples section for details on each file used in PACKMOL.Create a
molecule.namesfile with the following contents:1 Element# 2 Element# N Element# # N = number of atoms & Element# = the name of the element for the .xyz file.
For example:
1 H1
2 O1
3 H2 # for a water molecule
The molecule.names file will use the names when creating the .xyz file. This is used for every atom present in the system and requires unique names for each atom.
Important
Molecule names should be discrete for each atom present. This means that atom names cannot be the same across types. For example, a system which contains multiple hydrogen atoms (say for example, two types: HGA3 and HGP1 from CHARMM FF Docs) would need to have labels that separate between all hydrogens and not just each type. Therefore, hydrogen HGA3 could be shown as HGA3_1, HGA3_2, … with HGP1 labeled similarly. If this is not shown, the built .connect file will not display all bonds/angles/dihedrals correctly and the system will not run successfully.
Create a
molecule.paircoeffsfile with the following contents:pair_coeff 1 1 epsilon sigma pair_coeff 2 2 epsilon sigma pair_coeff NP NP epsilon sigma # where NP is the number of pair coefficient types.
Note
The number of pair coefficient types is not for every atom in the system, but for every atom type. For example, for a system with 11 atoms and 6 atom types will have 6 pair coefficient types.
Create a
molecule.bondcoeffsfile with the following contents:bond_coeff 1 k r bond_coeff 2 k r # k = force const & r = bond length bond_coeff NBT k r # NBT = number of bond types
Note
The number of bond coefficient types is not for every bond present in the system, but for every bond type. For example, a bond which contains the same length and force oefficient for two different O-H bonds will be considered the same type. See the examples section for details on bond types.
Create a
molecule.anglecoeffsfile with the following contents:angle_coeff 1 k theta angle_coeff 2 k theta # k = force const & theta = angle angle_coeff NAT k theta # NAT = number of angle types
Note
The number of angle coefficient types is not for every angle present in the system, but for every angle type. For example, an angle which contains the same force coefficient and angle for two different C-N-H angles will be considered the same type. See the examples section for details on angle types.
Create a
molecule.dihedralcoeffsfile with the following contents:dihedral_coeff 1 k n d w dihedral_coeff 2 k n d w # k = force const & n = int & d = dihedral angle & w = weighing factor dihedral_coeff NDT k n d w # NDT = number of dihedral types
Note
The number of dihedral coefficient types is not for every dihedral present in the system, but for every dihedral type. For example, a dihedral which contains the same force constant, integer, dihedral angle, and weighing factor for a C-N-C-H dihedral will be sindered the same type. See the examples section for details on dihedral types.
Create a
molecule.impropercoeffsfile with the following contents:improper_coeff 1 k X improper_coeff 2 k X # k = force const & X = improper angle improper_coeff NIT k X # NIT = number of improper types
Note
The number of improper coefficient types is not for every improper present in the system, but for every improper type. For example, an improper which contains the same force constant and improper angle for a C-N-N-O improper will be considered the same type. See the examples section for details on improper types.
Note
You may experience an error during the running PACKMOL step which shows another parameter for the impropers. This can be labeled as 0.0 unless otherwise listed in the force field data of the system.
Running PACKMOL
To run the PACKMOL system from the aforementioned configuration files, use the command
python molec_generator.py input output molname. For example:python molec_generator.py spce.connect spce.py spce. The.connectfile is required, andmolec_generator.pycreates the Python file,moleculename.pyautomatically.Once the
.pyfile is created, it is required to copy the file:cp molecule.py /path/to/My-Code-Collection/Util/general_system/molecules/molecule.py
Prior to building, a
molecule.inpfile must be created in the following specific format:{ "num_components":#, "nspec":[#, etc.], "tspec":["name (without .py)", etc.], "blength":#, "e_unit":["kcal", etc.], "f_unit":["kcal", etc.], "eo_unit":["kcal", etc.], "fo_unit":["kcal", etc.], "shift_f":1.0, "ff_type":["lj", etc.] }
Only one .inp file is made per system. For example, a 4M urea and water system will be formatted with the following:
{
"num_components":2,
"nspec":[330, 14],
"tspec":["tip3p-fb", "urea"],
"blength":22.5378973,
"e_unit":["kcal", "kcal"],
"f_unit":["kcal", "kcal"],
"eo_unit":["kcal", "kcal"],
"fo_unit":["kcal", "kcal"],
"shift_f":1.0,
"ff_type":["lj", "lj"]
}
To build the molecule, do so within the path which contains the
molecule.inpfile:> module load build > build.py < filename.inp
PACKMOL will automatically create several files which contain information for a LAMMPS simulation. The data.lmps file can simply be copied for the future LAMMPS simulation used.
Note
It is required to have the arrow in the command build.py < filename.inp facing the build.py file. If this direction is mirrored, you will lose your .inp file and the PACKMOL build process will not work!
The resulting data from PACKMOL can be used to cat into a LAMMPS in. file. For example, cat lmps.* >> in.nvt will write all the lmps. outputs into a file called in.nvt. You can edit the file with LAMMPS commands to create the system of interest for an NVT simulation (in this case). For LAMMPS files, in.name can be used for NVT, NVE, NpT, among many more types of simulations. Please see the LAMMPS docs for details on how to write and create LAMMPS simulations.
Examples
Examples are shown for a simplistic water model and an asymmetric Methylurea model.
TIP3P-FB Water Model
Consider a simplistic example of the TIP3P-FB water model. The path for this model can be found at kuhpc/thompson/work/a122k651/packmol/tip3p_fb.
The contents are as follows:
molec_generator.pytip3p_fb.anglecoeffstip3p_fb.bondcoeffstip3p_fb.connecttip3p_fb.inptip3p_fb.namestip3p_fb.paircoeffstip3p_fb.py
For the sake of simplicity, only the tip3p_fb.connect and tip3p_fb.bondcoeffs files are shown.
Connect File
# This file is a connectivity file for TIP3P/FB
3 atoms # O, H1, H2
2 bonds # all bonds, NOT types of bonds
1 angles # all angles
0 dihedrals
0 impropers
Coords
1 0.000000 0.000000 0.000000 # O
2 0.000000 0.000000 1.011800 # H1
3 0.961457 0.000000 -0.315182 # H2
Types
1 1 # Oxygen, atom 1, type 1
2 2 # Hydrogen1, atom 2, type 2
3 2 # Hydrogen2, atom 3, type 2
Charges
1 -0.84844 # O
2 0.42422 # H1
3 0.42422 # H2
Masses
1 15.9990 # O
2 1.0080 # H1
3 1.0080 # H2
Bonds
1 1 1 2 # first bond, type 1, O-H1
2 1 1 3 # second bond, type 1, O-H2
Angles
1 1 2 1 3 # first angle, type 1, H1-O-H2
The .connect for TIP3P-FB shows several key details:
The bonds listed within the
.connectfile state there are two bonds present. However, within theBondssubheading, there exists only one type of bond. This key difference is significant once the.bondsfile is made, which can be seen below.The two hydrogen atoms are considered the same type. However, within the
.connectfile, their masses are explicitly listed.Sections for
DihedralsandImpropersare not listed in the.connectfile. This is further shown in the list of files for the TIP3P-FB water model, where the.dihedralcoeffsand.impropercoeffsfiles are not present.
Bond Coefficients File
bond_coeff 1 553.000 1.0118 # O-H
From the .connect file, there are two listed bonds in the TIP3P-FB system. However, only one type of bond is defined. This is reflected via the .bondcoeffs file, which shows the types of bonds in the system.
Methylurea Model
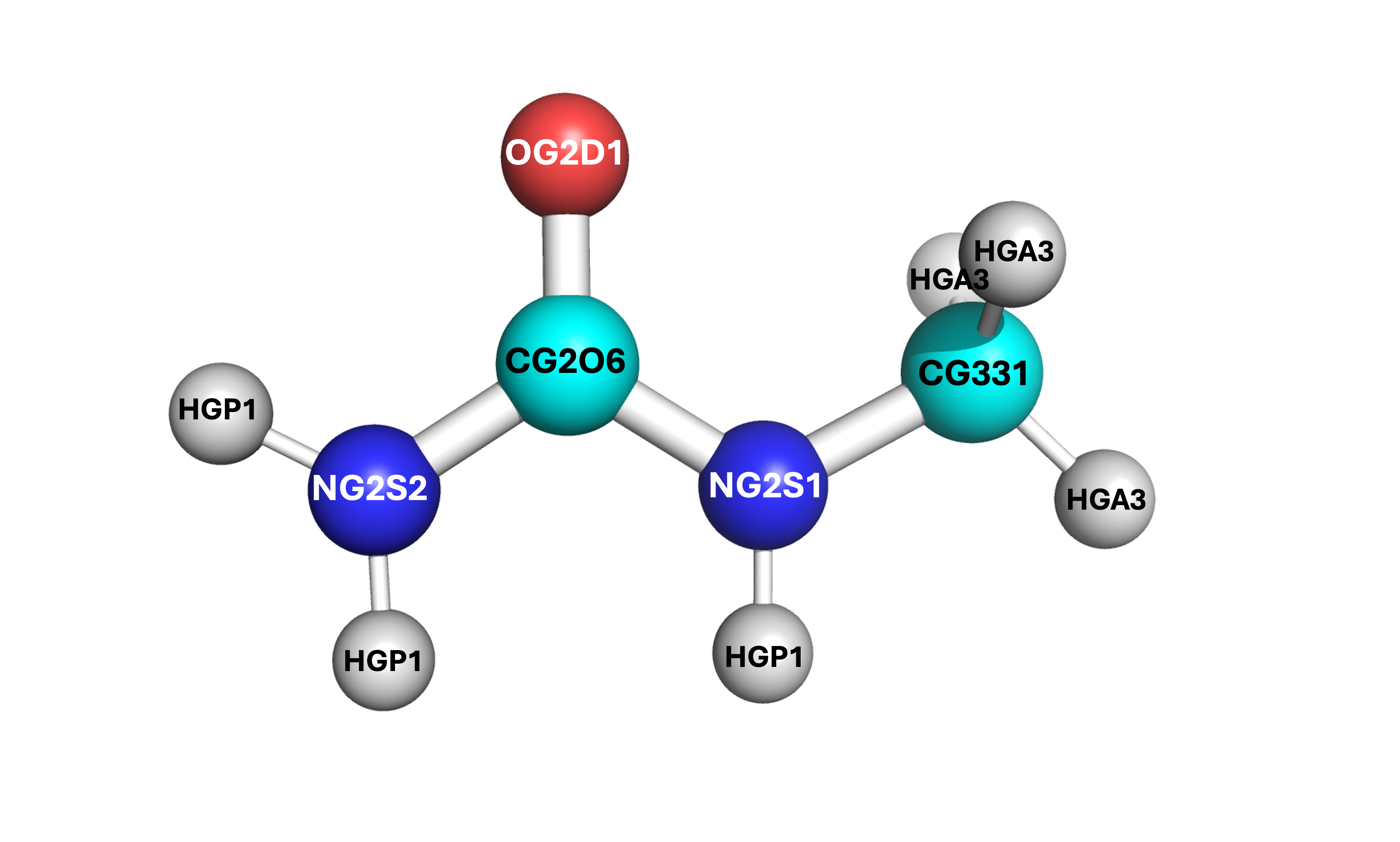
The methylurea model was developed via CGenFF from its structure obtained at https://pubchem.ncbi.nlm.nih.gov/compound/Methylurea. The structure is associated from the identifying atom labels given from the CHARMM force field data from CGenFF, which can be seen in the structure below. For details on its development into PACKMOL, see the section Developing Molecules for LAMMPS.
The files shown in the methylurea model are listed (see contents at kuhpc/thompson/work/a122k651/packmol/energy_mins/methylurea/starterfiles_methylurea):
methylurea.anglecoeffsmethylurea.connectmethylurea.impropercoeffsmethylurea.namesmethylurea.pymethylurea.bondcoeffsmethylurea.dihedralcoeffsmethylurea.inpmethylurea.paircoeffsmolec_generator.py
For the sake of simplicity, only the .connect, .anglecoeffs, and .dihedralcoeffs will be shown.
Connect File
# 1-MONOMETHYLUREA SYSTEM
11 atoms
10 bonds
15 angles
8 dihedrals
1 impropers
Coords
1 4.637224824258854 3.939505581798852 2.720061084395536 # CG331
2 4.234156216717911 4.221940890505587 4.05802642237323 # NG2S1
3 3.1658553620638044 3.5190398618536536 4.5548357700110875 # CG2O6
4 2.7872392741492495 3.8007105470089186 5.845925212783197 # NG2S2
5 2.582610268034431 2.677395841085764 3.8647055934269243 # OG2D1
6 3.260203074178855 4.483148645078783 6.405233040214911 # HGP1 (AMIDE SIDE)
7 2.011495906975985 3.3125153319773095 6.253272157273618 # HGP1 (AMIDE SIDE)
8 4.75773122119284 4.915999376212109 4.548650471430572 # HGP1 (METHYL SIDE)
9 5.505632660948109 4.569436036834264 2.4390591532339316 # HGA3
10 4.913210613313182 2.8674205690506347 2.6360109369635523 # HGA3
11 3.7921275781667827 4.142204318594125 2.0291791578934393 # HGA3
Types
1 1 # CG331
2 2 # NG2S1
3 3 # CG2O6
4 4 # NG2S2
5 5 # OG2D1
6 6 # HGP1
7 6 # HGP1
8 6 # HGP1
9 7 # HGA3
10 7 # HGA3
11 7 # HGA3
Charges
1 -0.011 # CG331
2 -0.342 # NG2S1
3 0.226 # CG2O6
4 -0.521 # NG2S2
5 -0.487 # OG2D1
6 0.296 # HGP1 (AMIDE SIDE)
7 0.296 # HGP1 (AMIDE SIDE)
8 0.273 # HGP1 (METHYL SIDE)
9 0.090 # HGA3
10 0.090 # HGA3
11 0.090 # HGA3
Masses
1 12.011 # CG331
2 14.007 # NG2S1
3 12.011 # CG2O6
4 14.007 # NG2S2
5 15.999 # OG2D1
6 1.008 # HGP1 (AMIDE SIDE)
7 1.008 # HGP1 (AMIDE SIDE)
8 1.008 # HGP1 (METHYL SIDE)
9 1.008 # HGA3
10 1.008 # HGA3
11 1.008 # HGA3
Bonds
1 1 3 2 # CG2O6 - NG2S1
2 2 3 4 # CG2O6 - NG2S2
3 3 3 5 # CG2O6 - OG2D1
4 4 1 2 # CG331 - NG2S1
5 5 1 9 # CG331 - HGA3 (same as 1 10, 1 11)
6 5 1 10 # CG331 - HGA3
7 5 1 11 # CG331 - HGA3
8 6 2 8 # NG2S1 - HGP1
9 7 4 6 # NG2S2 - HGP1 (same as 4 7)
10 7 4 7 # NG2S2 - HGP1
Angles
1 1 2 3 4 # NG2S1 - CG2O6 - NG2S2
2 2 2 3 5 # NG2S1 - CG2O6 - OG2D1
3 3 4 3 5 # NG2S2 - CG2O6 - OG2D1
4 4 2 1 9 # NG2S1 - CG331 - HGA3
5 4 2 1 10 # NG2S1 - CG331 - HGA3
6 4 2 1 11 # NG2S1 - CG331 - HGA3
7 5 9 1 10 # HGA3 - CG331 - HGA3
8 5 9 1 11 # HGA3 - CG331 - HGA3
9 5 10 1 11 # HGA3 - CG331 - HGA3
10 6 3 2 1 # CG2O6 - NG2S1 - CG331
11 7 3 2 8 # CG2O6 - NG2S1 - HGP1 (METHYL SIDE)
12 8 1 2 8 # CG331 - NG2S1 - HGP1 (METHYL SIDE)
13 9 3 4 7 # CG2O6 - NG2S2 - HGP1 (AMIDE SIDE)
14 9 3 4 6 # CG2O6 - NG2S2 - HGP1 (AMIDE SIDE)
15 10 7 4 6 # HGP1 - NG2S2 - HGP1 (AMIDE SIDE)
Dihedrals
1 1 4 3 2 1 # NG2S2 - CG2O6 - NG2S1 - CG331 (TRANS)
2 2 4 3 2 8 # NG2S2 - CG2O6 - NG2S1 - HGP1 (METHYL SIDE)
3 3 5 3 2 1 # OG2D1 - CG2O6 - NG2S1 - CG331 (CIS)
4 4 5 3 2 8 # OG2D1 - CG2O6 - NG2S1 - HGP1 (METHYL SIDE)
5 5 2 3 4 7 # NG2S1 - CG2O6 - NG2S2 - HGP1 (AMIDE SIDE)
6 6 5 3 4 6 # OG2D1 - CG2O6 - NG2S2 - HGP1 (AMIDE SIDE)
7 7 9 1 2 3 # HGA3 - CG331 - NG2S1 - CG2O6
8 8 9 1 2 8 # HGA3 - CG331 - NG2S1 - HGP1 (METHYL SIDE)
Impropers
1 1 3 2 4 5 # CG2O6 - NG2S1 - NG2S2 - OG2D1
There are differences in identification for each type of atom in methylurea which differs from that of the TIP3P-FB water system shown previously. It is vital to ensure that the atoms listed within the force field data are the same for the structure you create in PACKMOL. As previously emphasized, all bonds, angles, dihedrals, and impropers must be listed within the .connect file.
Angle Coefficiens File
angle_coeff 1 70.00 115.0 # NG2S1 - CG2O6 - NG2S2
angle_coeff 2 60.00 125.7 # NG2S1 - CG2O6 - OG2D1
angle_coeff 3 75.00 122.5 # NG2S2 - CG2O6 - OG2D1
angle_coeff 4 51.50 109.5 # NG2S1 - CG331 - HGA3
angle_coeff 5 35.50 108.4 # HGA3 - CG331 - HGA3
angle_coeff 6 60.00 120.0 # CG2O6 - NG2S1 - CG331
angle_coeff 7 40.00 121.5 # CG2O6 - NG2S1 - HGP1
angle_coeff 8 35.00 117.0 # CG331 - NG2S1 - HGP1
angle_coeff 9 50.00 120.0 # CG2O6 - NG2S2 - HGP1
angle_coeff 10 23.00 120.0 # HGP1 - NG2S2 - HGP1
As previously mentioned, all of the .parameter files for PACKMOL describe the types which are expressed in the .connect file. In this case, there are 15 total angles in the molecule but only 10 types, which are stated above. For data shown in the PACKMOL files, there may be terms which are present in the force field data that are not within the PACKMOL setup. For example, angle terms may have Urey-Bradley coefficients. These can be added once the data.lmps file is generated at the end of the PACKMOL process, and should not be inputted prior to running the molec_generator.py and build.py < file.inp steps. Only once the data.lmps file is created successfully may these terms be added.
Dihedral Coefficients File
dihedral_coeff 1 2.5000 2 180 0.0 # NG2S2 - CG2O6 - NG2S1 - CG331
dihedral_coeff 2 4.0000 2 180 0.0 # NG2S2 - CG2O6 - NG2S1 - HGP1
dihedral_coeff 3 0.9500 4 0 0.0 # OG2D1 - CG2O6 - NG2S1 - CG331
dihedral_coeff 4 0.0000 2 180 0.0 # OG2D1 - CG2O6 - NG2S1 - HGP1
dihedral_coeff 5 1.5000 2 180 0.0 # NG2S1 - CG2O6 - NG2S2 - HGP1
dihedral_coeff 6 1.4000 2 180 0.0 # OG2D1 - CG2O6 - NG2S2 - HGP1
dihedral_coeff 7 0.0000 3 0 0.0 # HGA3 - CG331 - NG2S1 - CG2O6
dihedral_coeff 8 0.0000 3 0 0.0 # HGA3 - CG331 - NG2S1 - HGP1
Dihedral coefficients require a weighing factor which is not present in the force field workup. These can be determined by reading the LAMMPS documentation (https://docs.lammps.org) to determine when weighing factors need to be non-zero values. Additionally, force field workups may include multiple lines which contain the same relevant atoms to a given dihedral. These can discern between cis and trans conformations and should be studied carefully prior to writing the PACKMOL .dihedralcoeffs file. It is recommended to observe the structure from its PBD file or from an online source to see the best optimized geometry.